機械学習でタンパク質を設計!?【Fold2Seq】
3つの要点
✔️ タンパク質の立体構造を細かく分割した単位立方体における二次構造の密度として表現し、各立体構造の表現をTransformer Encoderベースの深層学習モデルから獲得
✔️ タンパク質の立体構造とアミノ酸配列の両方を表現する埋め込みを学習することで、立体構造空間と配列空間という異なるドメインの関係性をとらえた
✔️ ベンチマークにおいて、SOTAであるRosettaDesignをパープレキシティー、配列復元率で超える性能を記録した
Fold2Seq: A Joint Sequence(1D)-Fold(3D) Embedding-based Generative Model for Protein Design
written by Yue Cao, Payel Das, Vijil Chenthamarakshan, Pin-Yu Chen, Igor Melnyk, Yang Shen
(Submitted on 24 Jun 2021)
Comments: ICML 2021
Subjects: Machine Learning (cs.LG); Biomolecules (q-bio.BM)
code:

本記事で使用している画像は論文中のもの、紹介スライドのもの、またはそれを参考に作成したものを使用しております。
はじめに
タンパク質とはアミノ酸が直鎖状に連結された物質であり、私たちの体を形作る重要な要素のうちの一つです。タンパク質は直線的な構造ではなく、それぞれ望ましい立体構造に折り畳まれることで生体的機能を発揮します。このようなタンパク質の折り畳みのことをフォールディングと呼びます。
近年では特定の構造を与えるようなアミノ酸配列を設計する問題が注目されており、この問題をInverse protein designと呼びます。この問題における課題として「探索する配列空間の広大さ」と「構造空間と配列空間の間のマッピングが難しい点」が挙げられます。
Inverse protein designにおける従来の研究はタンパク質の主鎖構造を与えて配列を設計するものが大多数であり、タンパク質フォールドを与えるものはほとんどありません。しかし、タンパク質の主鎖構造を与えた配列設計では、真新しい配列を設計しにくい点や設計される配列の多様性が損なわれる点が問題点として指摘されています。これはフォールドの方が主鎖構造よりも高次的な表現であるためであり、主鎖構造を限定すると暗黙的にアミノ酸配列の候補を狭めてしまうことを意味します。
そこで、今回紹介する論文では主鎖構造ではなくフォールディングを与えた配列設計に挑戦しています。タンパク質フォールドの多様性を担保した表現を獲得すること、フォールド空間と配列空間との間の複雑な関係性を克服することを主な目標としています。
Fold2Seq
タンパク質フォールドの表現方法
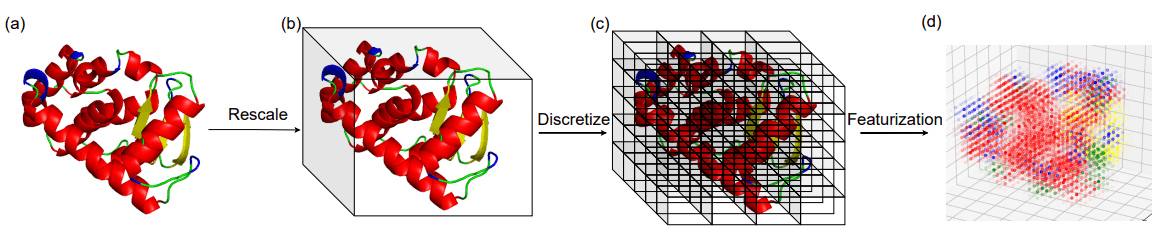
タンパク質におけるフォールドとは局所的な二次構造要素(SSE)の立体的配置のことを指します。筆者らはタンパク質の立体構造の占める3次元空間を単位立方体で分割し、各立方体における二次構造要素(SSE)の密度をフォールドの表現として採用しています。
考慮された二次構造要素はヘリックス、βストランド、ループ、ベンド・ターンの4種類です。タンパク質のあるアミノ酸残基$j$が単位立方体$i$に与える影響度をガウス分布で表現し、各二次構造要素に対応するOne-Hot表現$t_j$を掛け合わせて以下のような特徴ベクトルが得られます。
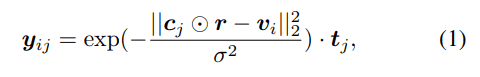
この特徴ベクトルをアミノ酸残基に関して足し合わせることにより、ある立方体におけるタンパク質二次構造の密度を各アミノ酸残基由来の混合ガウス分布として表現できます。
フォールドの表現を計算する際のイメージは以下の図に示す通りです。
アーキテクチャと損失関数
Fold2Seqのアーキテクチャ及び損失関数の関係は以下に示す通りです。
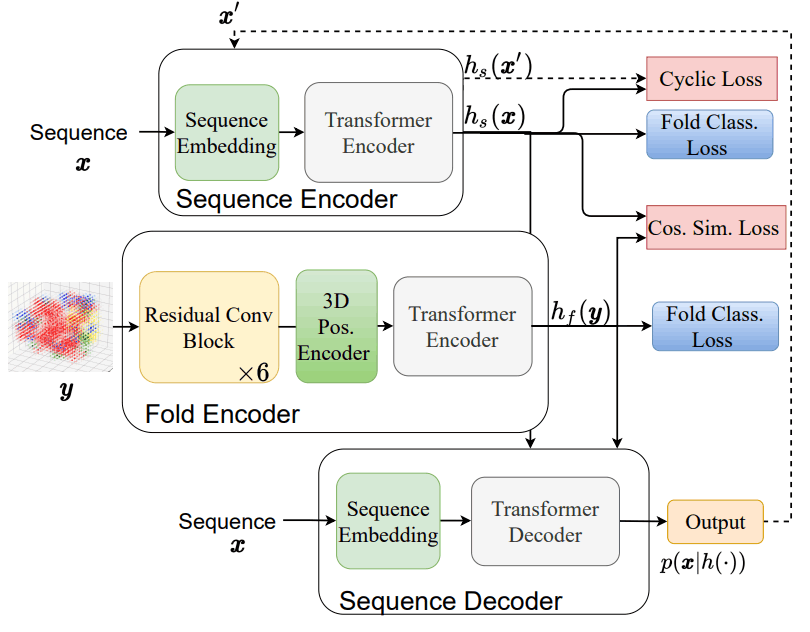
このアーキテクチャは以下の3つの要素から成り立っています。
- 配列エンコーダー$h_s$:アミノ酸配列を潜在空間に落とし込む役割を果たしており、従来のTransformer Encoderを採用しています。
- フォールドエンコーダー$h_f$:タンパク質フォールドを潜在空間に落とし込む役割を果たしており、3次元畳み込み層で構成された残差ニューラルネットワークを採用しています。
- 配列デコーダー$p(x|h(.))$:潜在空間からアミノ酸配列を出力する役割を果たしており、従来のTransformer Decoderを採用しています。
Fold2Seqの学習にはJoint Embedding Learningの枠組みが取り込まれており、立体構造と配列のような離れたドメイン間の関係性をうまくとらえるためには二つのドメイン内損失(intra-domain)と一つのドメイン間損失(cross-domain)が必要であるとしています。
ドメイン内損失とは生体内で同じ機能を持つ配列同士ないしフォールド同士を潜在空間上で近くに配置する損失であり、ドメイン間損失とは生体内で同じ機能を持つ配列とフォールドを潜在空間上で近くに配置する損失です。
Fold2Seqの全体の損失は以下に示す通りです。各項について以下で詳しく見ていきます。
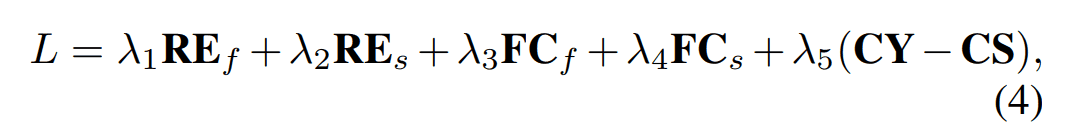 $RE_f, RE_s$
$RE_f, RE_s$
オートエンコーダモデルでのシンプルな再構成損失です。フォールドを入力して配列を再構成する損失を$RE_f$、入力配列を再構成する損失を$RE_s$として区別しています。
$FC_f, FC_s$
先述したドメイン内損失に該当する損失です。配列エンコーダーの出力$h_s(x)$とフォールドエンコーダーの出力$h_f(y)$をそれぞれ長さ方向に平均した特徴ベクトルから各タンパク質フォールドのクラスラベルを分類します。このタスクにおける交差エントロピー損失を配列とフォールドそれぞれに対して定義し、$FC_s, FC_f$としています。
この分類タスクを入れることによって同じクラスに属する配列とフォールドは似た潜在表現となるため、ドメイン間損失としての役割も果たすことになります。
$CS$
配列エンコーダーの出力$h_s(x)$とフォールドエンコーダの出力$h_f(y)$に対してコサイン類似度を計算します。対応するフォールド部位と配列部位の間で潜在表現が近くなるようにする効果があります。
$CY$
CycleGANのcyclic lossを参考にした項です。フォールドエンコーダーと配列デコーダーを用いて生成した配列を配列エンコーダーに入力して潜在表現$h_s(x')$を得ます。そして元の配列の潜在表現$h_s(x)$とのL2距離を損失とすることで生成した配列が元の配列から遠く離れないように制限します。
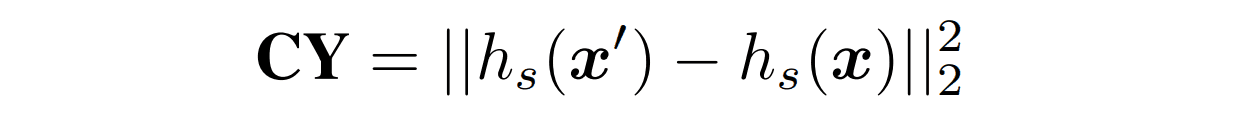
ネットワークアーキテクチャと損失関数を図示すると以下のようになりまていあんしていモデルの訓練と配列の生成
上述した損失関数を用いてモデルを学習する際に、配列エンコーダーとフォールドエンコーダーを同時に学習してしまうと、フォールドエンコーダーの学習が進行しないという現象が起きたため、筆者らは以下のような二段階の訓練手法を提案しています。
- $L_1=\lambda_2 RE_s + \lambda_4 FC_s$により配列エンコーダーと配列デコーダーを学習する
- 配列エンコーダーの重みを固定し、$L_2=\lambda_1 RE_f + \lambda_3 FC_f + \lambda_5 (CY - CS)によりフォールドエンコーダーと配列デコーダーを学習する$
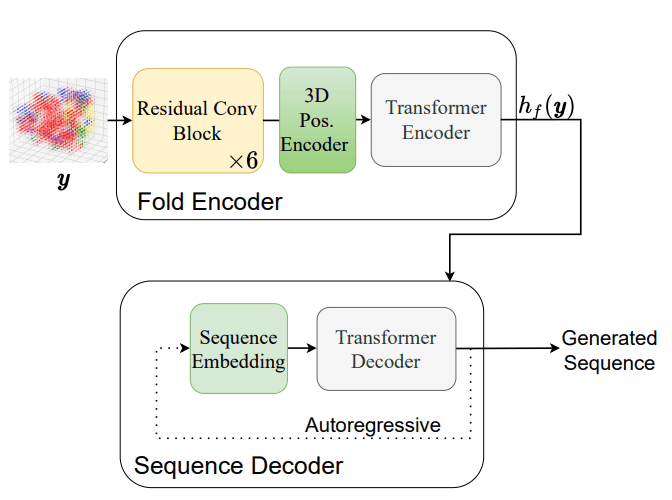
モデルを学習させた後に、実際にタンパク質フォールドから配列を生成する際には従来のTransformerによる自己回帰的な推論を行っています。配列を生成する際の手順は以下の図に示す通りです。
設計した配列の評価
Fold2Seqを用いて生成した配列の質を評価するため、構造レベルの評価指標を4つ定義しています。今回はその中の二つに絞って紹介します。
元論文では以下のような配列ドメイン上での評価指標だけでなく、構造ドメインでの評価指標も用いています。興味のある方は参照してみてください。
アミノ酸残基ごとのパープレキシティー
タンパク質フォールド$i$を構成する構造の集合$S_i$において、各構造に属する配列におけるパープレキシティーを計算したものです。フォールド$i$に対して計算されるパープレキシティーの値は以下のように定義されます。小さいほど良い指標です。
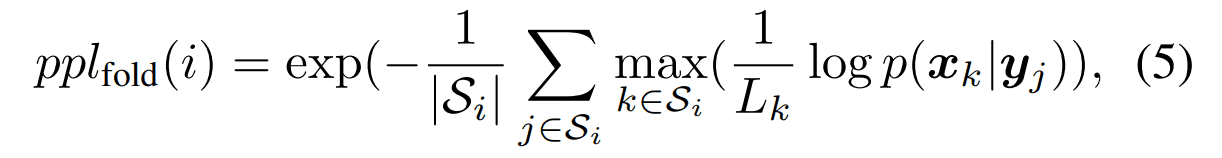
カバレッジ
フォールド$i$から代表例として一つの構造を選択し、その構造から生成される配列が元のフォールド$i$に属する配列のうちどの程度を生成できたか評価する指標です。ある配列が適切に生成されたかを判定する基準は配列類似度が30%以上としています。
具体的な算出方法は以下の通りです。ある構造kから生成された配列の集合を$G_k$としています。
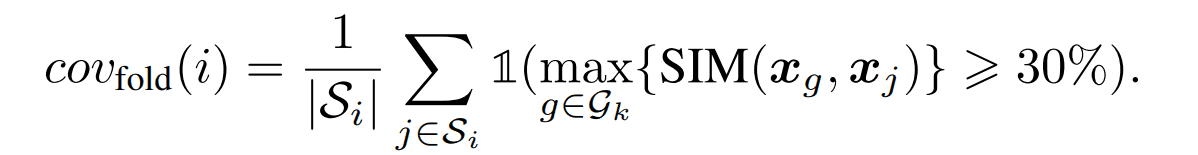
結果
Fold2SeqをCATH4.2(タンパク質の立体構造情報と配列情報を含むベンチマーク)に関して学習および評価した結果を簡単に紹介します。
下の表はアミノ酸残基ごとのパープレキシティーに関して評価した結果です。参考としてアミノ酸を一様分布からサンプリングした際のパープレキシティー(uniform)と、UniRef50の全アミノ酸配列を用いて計算したパープレキシティー(natural)が記載されています。
また、cVAE及びgcWGANは深層生成モデルを用いてタンパク質フォールドからアミノ酸配列を設計する比較手法となっており、Graph_transは主鎖構造をグラフ構造としてモデルに入力し、配列を設計する比較手法となっています。
訓練データと構造に被りが存在するテストデータセット(ID Test)と被りが存在しないテストデータセット(OD Test)において性能を比較しています。全体の傾向としてOD Testの方がパープレキシティーが高くなっています。
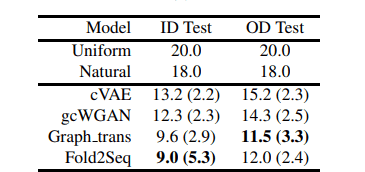
上の結果から、Fold2Seqは深層生成モデルを用いた他手法を超える性能を記録し、主鎖構造のような高解像度の構造情報を与えた場合と同程度の性能を出すことができていることがわかります。
また、下の表はカバレッジに関して評価した結果です。テストデータセットに含まれるフォールドを、そのフォールドに属する配列の本数が3本以上かどうかで分け、それぞれに関してカバレッジを計算しています。
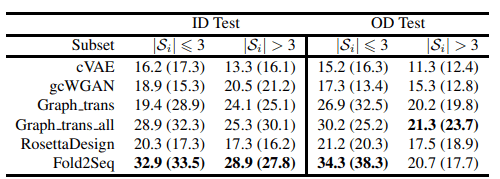
上の結果から、Fold2Seqは既存手法と比較してタンパク質フォールド内の配列多様性をより良くとらえられていることがわかります。RossettaDesignなどの物理学的原理に基づいた手法よりも優れた性能を出している点に注目です。
この論文では他にも設計に要する時間や、アミノ酸残基が欠失した構造の入力に対するロバスト性などの観点からFold2Seqの優位性を主張し、ツールとしての実用性にまで踏み込んで議論していました。
まとめ
いかがだったでしょうか。機械学習というと画像や自然言語などの分野で発展してきたという印象がありますが、生物学のような一見関係のない分野にまで機械学習が応用されているのには驚きです。
最近では配列からタンパク質の立体構造を予想するAlphaFold2が大きな注目を集めており、立体構造から配列を予測するという逆の方向性についても今後研究が加速しそうです。
生体内で期待通りの働きをしてくれるタンパク質を設計できる未来はそう遠くないのかもしれません。






この記事に関するカテゴリー




