用机器学习设计蛋白质

本文所使用的图片要么来自论文、介绍性幻灯片,要么是参考这些图片制作的 。
简介
蛋白质是由氨基酸的线性链组成的,是我们身体最重要的组成部分之一。蛋白质没有线性结构,而是被折叠成所需的三维结构,以发挥其生物功能。蛋白质的这种折叠被称为折叠。
近年来,设计一个氨基酸序列以给出一个特定结构的问题引起了人们的关注,这个问题被称为逆向蛋白质设计。这个问题的挑战在于需要探索的序列空间的广阔性以及结构空间和序列空间之间的映射的困难。
以前的逆向蛋白质设计工作大多是基于蛋白质的主链结构,而很少是基于蛋白质的折叠。然而,有人指出,基于蛋白质主链结构的序列设计是有问题的,因为它使设计新的序列变得困难,并减少了可设计的序列的多样性。这是因为折叠是比主链结构更高阶的表示,限制主链结构隐含地缩小了候选氨基酸序列的范围。
因此,在这里提出的论文中,我们试图设计出给予折叠而不是主链结构的序列。主要目标是获得一个确保多样性的蛋白质折叠的代表,并克服折叠空间和序列空间之间的复杂关系。
褶皱的二维码 (Fold2Seq)
如何表示一个蛋白质折叠
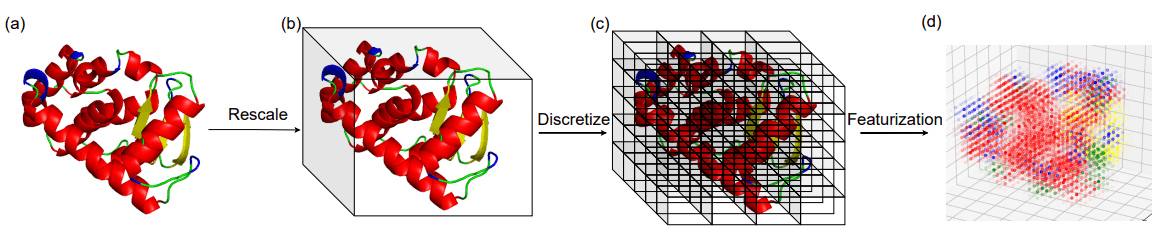
蛋白质中的折叠是局部二级结构元素(SSE)的三维排列。作者将蛋白质结构所占据的三维空间划分为单元立方体,并将每个立方体中二级结构元素(SSE)的密度作为折叠的代表。
考虑的四种类型的二级结构元素是螺旋、β-链、环和弯曲-转弯。蛋白质的某个氨基酸残基$j$对单位立方体$i$的影响用高斯分布表示,并与每个二级结构元素对应的一热表示法$t_j$相乘,得到以下特征向量。
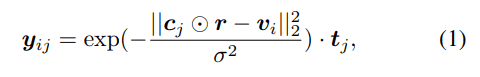
通过将这些相对于氨基酸残基的特征向量加在一起,蛋白质二级结构在立方体中的密度可以表示为由每个氨基酸残基衍生的混合高斯分布。
下图显示了用于计算折线表示法的图像。
结构和损失函数
Fold2Seq的结构和损失函数之间的关系如下所示。
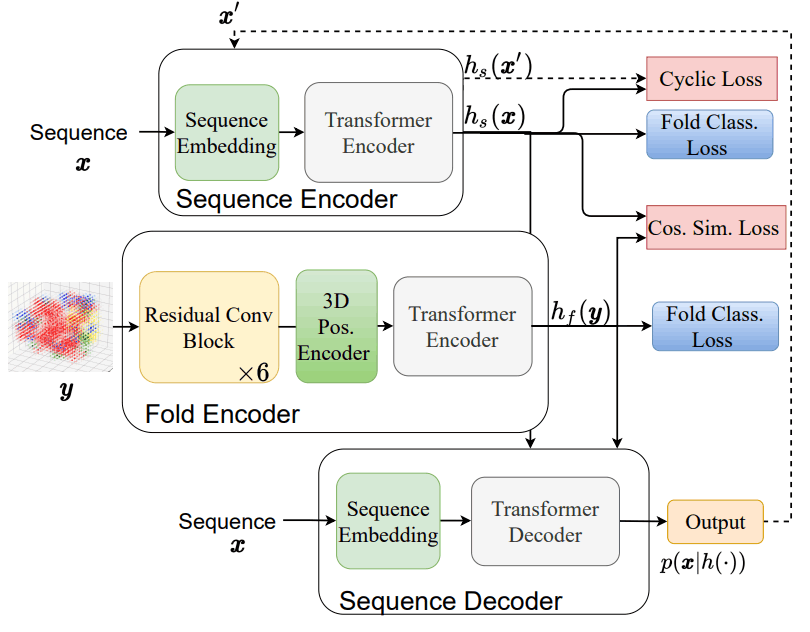
这个架构由三个要素组成
- 序列编码器$H_S$:负责将氨基酸序列投放到潜伏空间,是一个传统的转化器编码器。
- 折叠编码器$h_f$:它负责将蛋白质折叠投放到潜伏空间,并采用了由三维卷积层组成的残差神经网络。
- 阵列解码器 $p(x|h(.))美元:负责从潜伏空间输出氨基酸序列,是一个传统的变形器解码器。
Fold2Seq训练采用了联合嵌入学习的框架,即需要两个域内损失和一个跨域损失来成功捕捉远距离域之间的关系,如三维结构和序列。域)是成功捕捉远距离域之间关系的必要条件,如构象和序列。
域内损失是指将体内具有相同功能的序列或褶皱在潜伏空间中相互靠近的损失,而域间损失是将体内具有相同功能的序列和褶皱在潜伏空间中相互靠近的损失。
Fold2Seq的整体损失如下所示。下面将更详细地介绍每个术语。
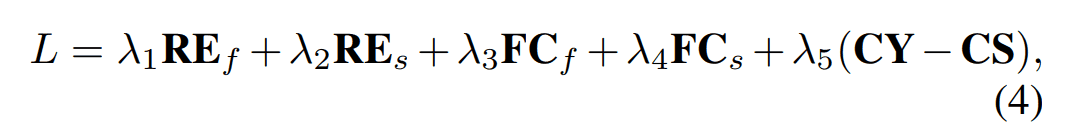 $RE_f, RE_s$。
$RE_f, RE_s$。
这是自动编码器模型中一个简单的重建损失。我们把重建一个有输入褶皱的序列的损失区分为$RE_f$和重建一个输入序列的损失为$RE_s$。
$FC_f, FC_s$。
这种损失与上述的域内损失相对应。我们通过对阵列编码器的输出$h_s(x)$和折叠编码器的输出$h_f(y)$在长度方向上分别进行平均得到的特征向量对每个蛋白质折叠的类别标签进行分类。这个任务中的交叉熵损失被定义为每个数组和折叠的$FC_s, FC_f$。
纳入这一分类任务也是一种领域间的损失,因为属于同一类别的序列和褶皱会有类似的潜在表示。
$CS$。
对阵列编码器的输出$h_s(x)$和折叠编码器的输出$h_f(y)$计算余弦相似度。其效果是使潜伏表征在相应的折叠和阵列位点之间更加紧密。
$CY$。
本节是基于CycleGAN的周期性损失。由褶皱编码器和阵列解码器产生的阵列被输入到阵列编码器,以获得潜伏表示$h_s(x')$。然后,与原始序列的潜在代表$h_s(x)$的L2距离被用作损失,以限制生成的序列与原始序列相差不大。
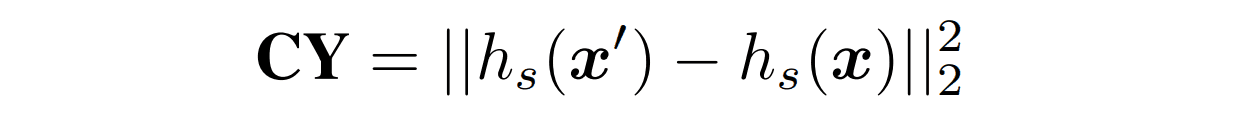
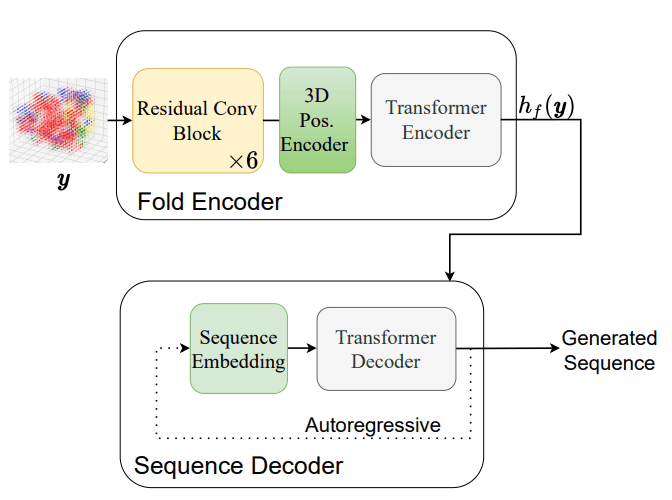
网络结构和损失函数如下图所示,它显示了模型训练和序列生成。
当使用上述损失函数训练模型时,如果阵列编码器和折叠编码器同时训练,则折叠编码器的训练不会取得进展,因此作者提出了两阶段的训练方法,如下所示
- $L_1= RE_s + λ_4 FC_s$ 来学习阵列编码器和阵列解码器
- 固定阵列编码器的权重,通过$L_2=RE_1 RE_f + λ_3 FC_f + λ_5 (CY - CS) $训练折叠编码器和阵列解码器。
训练完模型后,使用传统的自回归推理与Transformer进行蛋白质折叠序列的实际生成。序列生成所涉及的步骤如下图所示。
对所设计的序列进行评估
为了评估使用Fold2Seq生成的序列的质量,我们定义了四个结构级指标。在这篇文章中,我们将重点讨论其中的两个。
在最初的论文中,我们不仅使用了序列域的度量,还使用了结构域的度量,如下图所示。如果你有兴趣,请参考它。
每个氨基酸残基的困惑度
在构成蛋白质折叠的结构集$S_i$中,属于每个结构的序列的困惑度被计算。对一个褶皱$i$计算的紫度值定义如下。数值越小,指数越好。
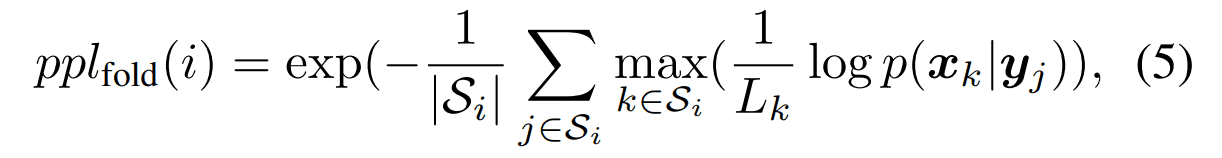
覆盖范围
这是一个评估从褶皱$i$中选取的结构作为代表例子所产生的序列能够产生属于原始褶皱$i$的序列的程度的措施。确定一个序列是否正确生成的标准是序列相似度至少为30%。
具体计算方法如下。我们用$G_k$表示由某个结构k产生的数组集合。
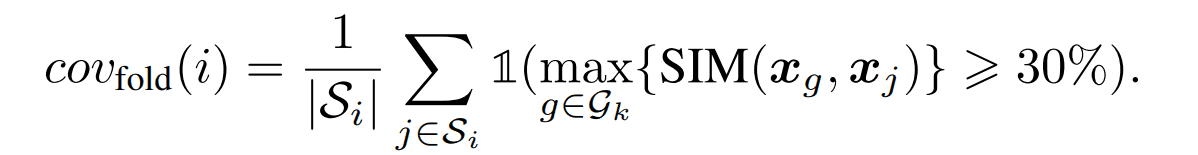
结果
我们简要介绍了针对CATH4.2(一个包括蛋白质构象和序列信息的基准)训练和评估Fold2Seq的结果。
下表显示了对每个氨基酸残基的紫度的评价结果。作为参考,它显示了通过从均匀分布的氨基酸中抽样计算的紫度(uniform)和使用整个UniRef50氨基酸序列计算的紫度(natural)。
cVAE和gcWGAN是使用深度生成模型从蛋白质褶皱中设计氨基酸序列的比较方法,而Graph_trans是通过将主链结构作为图结构输入模型而设计序列的比较方法。
我们比较了训练数据与结构重叠的测试数据集(ID测试)和没有重叠的测试数据集(OD测试)的性能。总的趋势是,OD测试的困惑度更高。
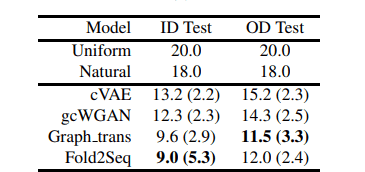
从上述结果可以看出,Fold2Seq的表现优于其他使用深度生成模型的方法,并且在给定高分辨率的结构信息(如主链结构)时,能够有同样的表现。
下表还显示了覆盖率评估的结果。测试数据集中的褶皱被划分为属于该褶皱的序列数量(3个或更多),并计算每个序列的覆盖率。
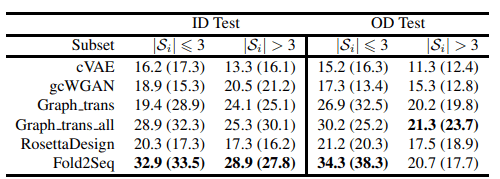
上述结果表明,Fold2Seq比现有的方法更好地捕捉了蛋白质折叠内的序列多样性,注意到它比RossettaDesign等基于物理学的方法表现更好。
论文还论证了Fold2Seq在设计时间和对缺乏氨基酸残基的结构输入的鲁棒性方面的其他优势,并继续讨论其作为一种工具的实用性。
摘要
你怎么看?机器学习已经在成像和自然语言等领域得到发展,但看到它如何被应用于生物学等看似不相关的领域,令人感到惊讶。
最近,从蛋白质序列预测其三维结构的AlphaFold2吸引了大量的关注,而从三维结构预测序列这一相反方向的研究在未来可能会加速。
也许用不了多久,我们就能设计出完全按照我们所期望的那样在体内做的蛋白质。



与本文相关的类别



![打开DNA的语言[DNABERT]](https://aisholar.s3.ap-northeast-1.amazonaws.com/media/December2021/dnabert-min-520x300.png)
